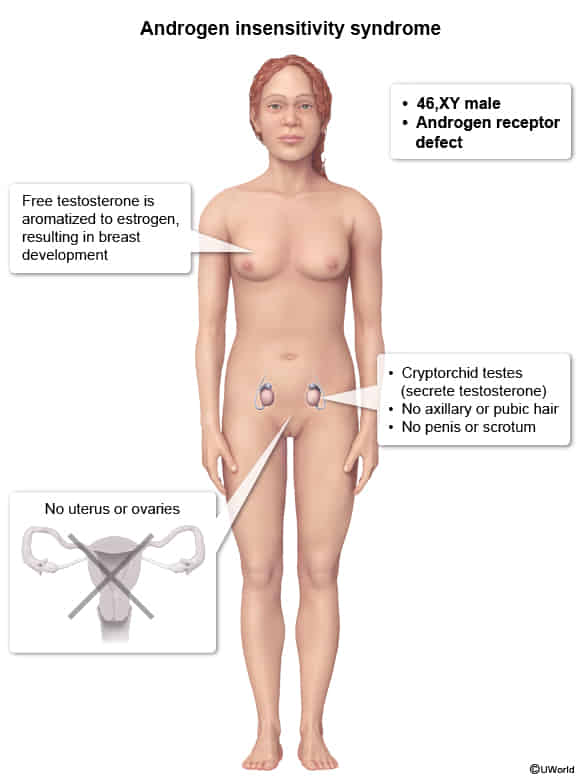
Androgen insensitivity syndrome
| Feature | Müllerian agenesis | Androgen insensitivity syndrome | 5-alpha reductase deficiency |
|---|---|---|---|
| Karyotype | 46,XX | 46,XY | 46,XY |
| Pathogenesis | Absent or hypoplastic müllerian ducts | Androgen resistance due to X-linked androgen receptor mutation | Deficient conversion of testosterone to dihydrotestosterone (DHT) |
| Hormone levels | Normal estrogen & testosterone | ↑ Testosterone & estrogen (aromatization) | ↑ Testosterone, ↓ DHT |
| Reproductive organs | Absent or rudimentary uterus/upper vagina, normal ovaries | No uterus/upper vagina; cryptorchid testes | No uterus/upper vagina; may have cryptorchid or descended testes |
| Breasts | Normal | Normal | Minimal to absent (Testosterone is enough to suppress breast) |
| Axillary & pubic hair | Normal | Minimal to absent | Sparse to normal (varies) |
| External genitalia | Normal female | Female appearance | Often ambiguous at birth, may virilize at puberty |
| Wolffian structures | Absent | Absent | Partial development |
| Gender identity | Female | Usually female | May change to male at puberty |
| Timing of diagnosis | Usually at adolescence (primary amenorrhea) | May be diagnosed at birth, puberty, or adulthood | Often at birth (ambiguous genitalia) or puberty |
| Risk of malignancy | No increased risk | ↑ Risk of gonadal tumors after puberty | ↑ Risk of gonadal tumors |
Klinefelter syndrome
Epidemiology
- One of the most common causes of male hypogonadism
Etiology
- Associated with advanced maternal age
Pathophysiology
- 47,XXY (rarely 48,XXXY or 48,XXYY)
- Presence of a Barr body (inactivated X chromosome)
- Testicular dysgenesis leads to:
- Seminiferous tubules dysgenesis → loss of Sertoli cells → ↓ inhibin B → ↑ FSH
- Leydig cell dysfunction → ↓ testosterone → ↑ LH
- Both ↑ LH and ↑ FSH lead to increased conversion of testosterone to estrogen.
Clinical features
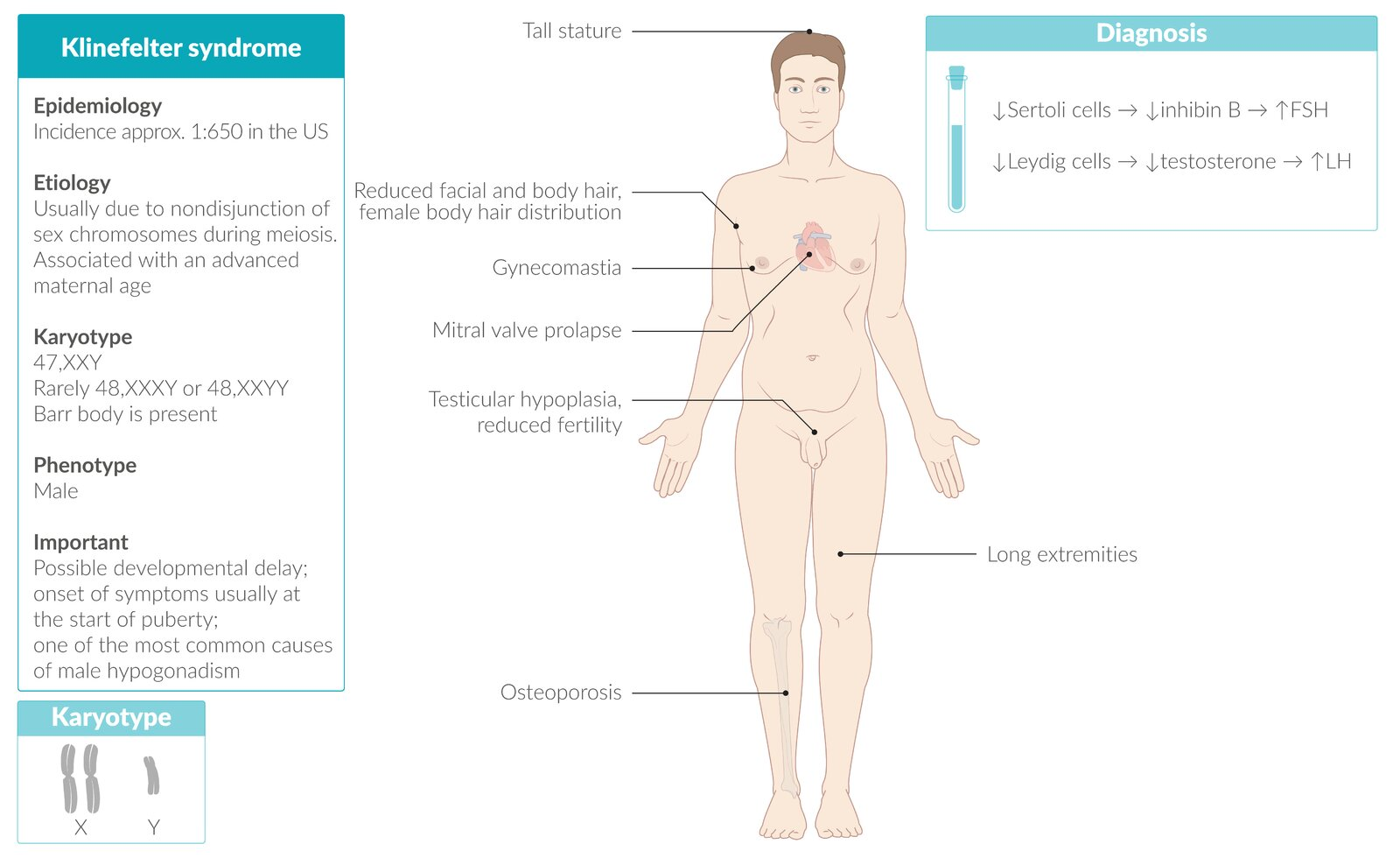
- Eunuchoid growth pattern: tall, slim stature with long extremities (Growth plate closure is delayed )
- Gynecomastia
- Testicular atrophy
- Possible developmental delay
- Neurocognitive dysfunction (impaired executive function and memory, decreased intelligence)
- Associated disorders
- Mitral valve prolapse
- Increased risk of breast and testicular cancer
- Due to increased estrogen
Turner syndrome
Pathophysiology
- Chromosomal nondisjunction → chromosome X monosomy/mosaicism → impaired ovarian development → malfunctioning streak gonads with connective tissue instead of normal germ cells → estrogen and progesterone deficiencies
- The absence of a second X chromosome disrupts X-linked survival signals, causing primordial germ cells to undergo accelerated apoptosis, leading to near-total germ cell loss by birth. Without germ cells, ovarian structures (e.g., follicles) fail to develop, and regressing gonadal ridges are replaced by fibrous connective tissue, forming non-functional streak gonads.
- Karyotype
- Meiotic nondisjunction (most often in paternal gametes) → complete sex chromosomal monosomy (45,XO; no Barr body)
- Barr body: The inactive X chromosome present in all female somatic cells. Appears as a small, dark-staining spot at the periphery of the nucleus. Consists of tightly-packed, transcriptionally-inactive, heterochromatin.
- Mitotic nondisjunction of an embryonic cell → sex chromosomal mosaicism (45,XO/46,XX) → mild phenotypic expression
- Meiotic nondisjunction (most often in paternal gametes) → complete sex chromosomal monosomy (45,XO; no Barr body)
Clinical features
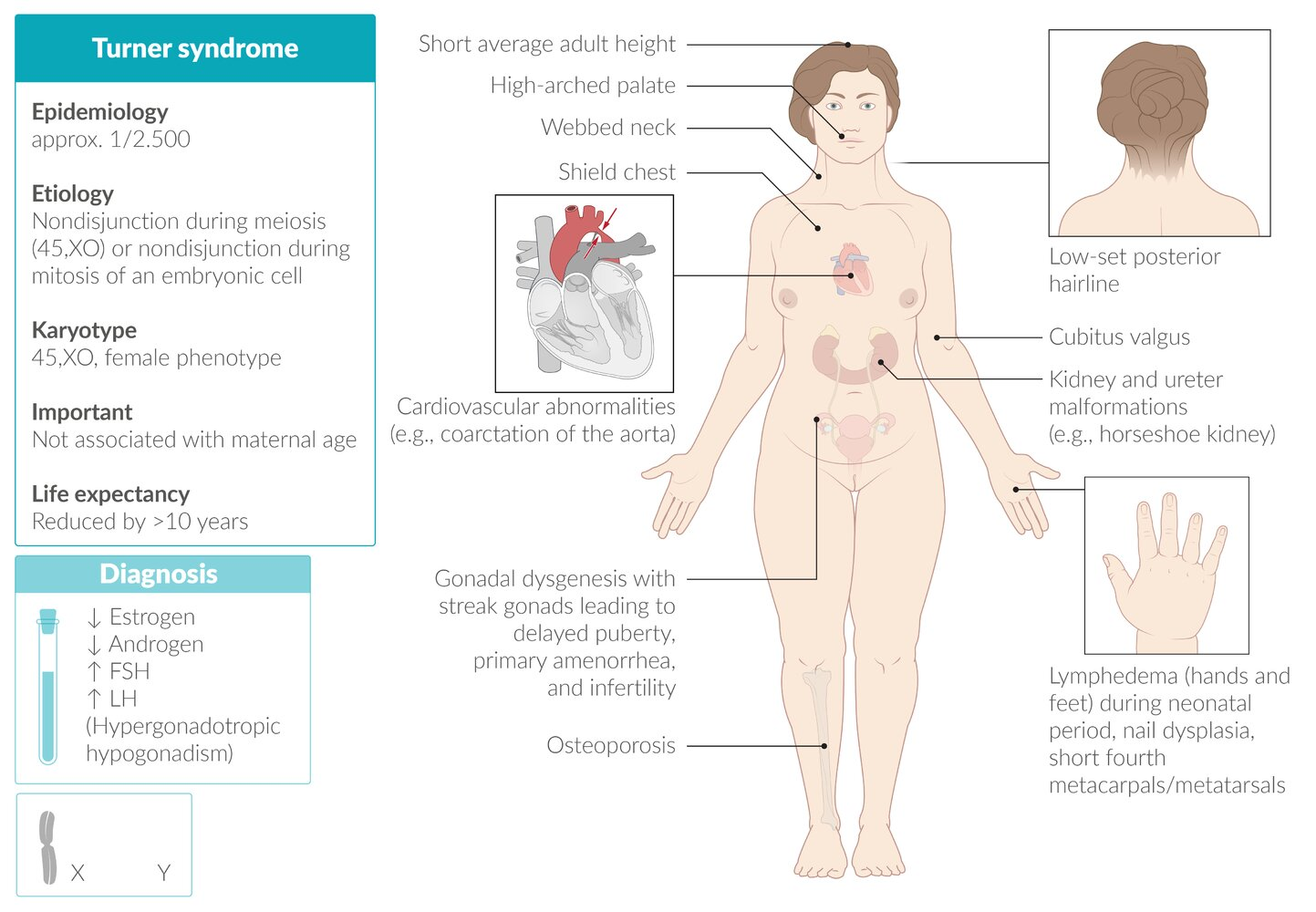
- Lymphatic system abnormalities
- Cystic hygroma
- a congenital lymphatic cyst (macrocystic lymphangioma) in the posterior triangle of the neck caused by malformation and obstruction of the fetal lymphatic system
- Present at birth as a soft, compressible, painless, posterior triangle neck mass
- Can cause dysphagia or airway compromise
- Lymphedema of the hands and feet in the neonatal period
- Cystic hygroma
- Musculoskeletal findings
- Short stature: due to the presence of only one copy of the SHOX (short stature homeobox) gene, normally located on the X chromosome
- Scoliosis are common
- Shield chest: broad chest with widely spaced nipples
- Webbed neck: skin folds along the side of the neck between the mastoid process and the acromion
- Cubitus valgus
- Short fourth metacarpals/metatarsals, nail dysplasia
- High arched palate
- Low-set posterior hairline
- Osteoporosis and pathologic fractures
- Cardiovascular abnormalities
- Bicuspid aortic valve: increased risk of premature aortic stenosis and/or insufficiency
- As a result of valve calcification.
- Coarctation of the aorta with brachial-femoral delay
- Aortic dissection and rupture
- Hypertension (even in children)
- Bicuspid aortic valve: increased risk of premature aortic stenosis and/or insufficiency
- Other disorders
- Gonadoblastoma (especially in patients with 45,XO/46,XY mosaicism)
- Malformations of the kidney and ureters (especially horseshoe kidney)
- Hashimoto thyroiditis
- Type 2 diabetes mellitus
Tip
Most patients with Turner syndrome have normal intelligence.
Kallmann syndrome
Etiology
Hypogonadotropic hypogonadism with hyposmia/anosmia
Pathophysiology
- Defective migration of GnRH-releasing neurons from the olfactory bulbs to the hypothalamic preoptic nuclei → ↓ GnRH secretion and underdevelopment of the olfactory bulbs
- ↓ GnRH → ↓ pituitary secretion of FSH and LH → ↓ testosterone in male individuals and ↓ estrogen in female individuals
Clinical features
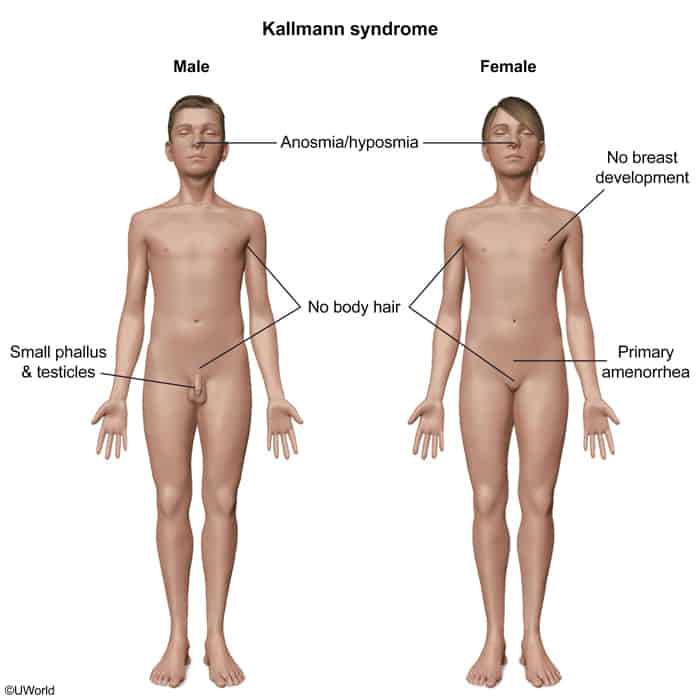
- Anosmia or hyposmia
- Infertility
Androgen insensitivity syndrome
- Pathophysiology
- Dysfunction of the AR in the hypothalamus and pituitary leads to loss of feedback inhibition of gonadotropin-releasing hormone (GnRH), FSH, and LH. This results in the following hormonal findings:
- GnRH induces increased LH secretion, which leads to increased testosterone production in the testes.
- FSH secretion is increased by GnRH but suppressed by inhibin from the seminiferous tubules and is often normal.
- Estrogen, which is derived by aromatization of testosterone, may be normal or elevated.
- Dysfunction of the AR in the hypothalamus and pituitary leads to loss of feedback inhibition of gonadotropin-releasing hormone (GnRH), FSH, and LH. This results in the following hormonal findings:
- Diagnostics
- Clinical presentation
- Before puberty: ↑ testosterone
- After puberty: ↑ LH, ↑ estrogen, and normal/↑ testosterone levels (no virilization)
- Genetic testing