Epidemiology
- Peak incidence
- ∼ 40 years of age (symptom onset usually between 20 and 50 years of age)
- One of the most common hereditary diseases of the brain
Etiology
- Increased number of CAG repeats (trinucleotide or triplet repeat expansion) in the huntingtin gene on chromosome 4 (most likely due to DNA polymerase dysfunction) results in the expression of an altered huntingtin protein.
- Huntingtin is physiologically expressed throughout the CNS, but its exact function is not known.
- Autosomal dominant
- Anticipation: increase in the number of CAG repeats in subsequent generations
- In genetics, the term “anticipation” refers to the increasing severity and/or increasingly earlier manifestation of a disease from generation to generation. This phenomenon is observed in HD.
Pathophysiology
Tip
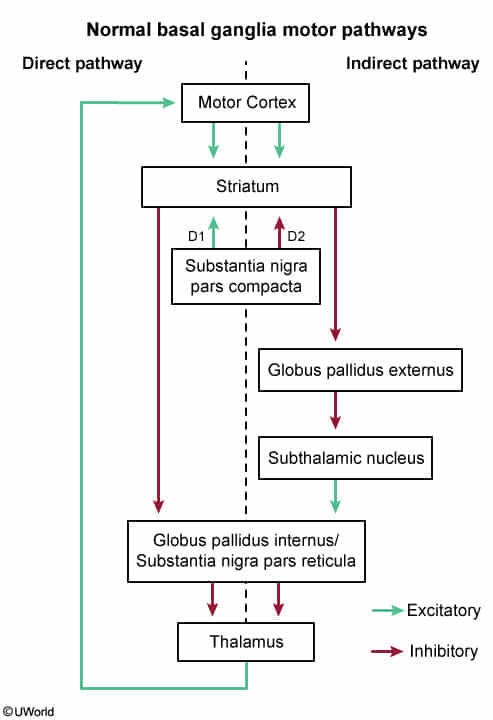
- Leads to atrophy of the caudate and putamen (striatum).
- Neurotransmitter changes: ↓ GABA, ↓ ACh, ↑ Dopamine.
- Toxic Gain of Function: Mutated huntingtin protein aggregates inside neurons, causing toxicity.
- Neuronal Death: Selective atrophy of the striatum (caudate nucleus and putamen).
- Loss of spiny GABAergic neurons is the hallmark.
- Excitotoxicity: Potential mechanism involving excessive activation of NMDA receptors by glutamate, leading to neuronal cell death.
- Neurotransmitter Changes
- ↓ GABA (gamma-aminobutyric acid): Loss of inhibition leads to hyperkinetic movement.
- ↓ ACh (acetylcholine).
- ↑ Dopamine: Relative excess contributes to chorea.
Mnemonic

Clinical features
- Initial stages
- Movement dysfunction
- Chorea: involuntary, sudden, irregular, nonrepetitive, arrhythmic movements of the limbs, neck, head, and/or face
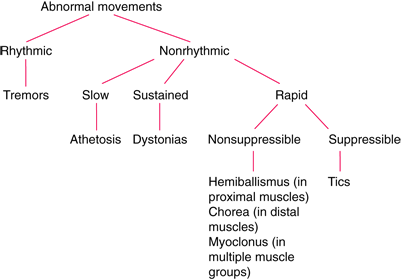
- Athetosis: involuntary, writhing movements, particularly of the hands and fingers
- Chorea: involuntary, sudden, irregular, nonrepetitive, arrhythmic movements of the limbs, neck, head, and/or face
- Movement dysfunction
- Advanced stages
- Movement dysfunction
- Hypokinetic motor symptoms: dystonia, rigidity, bradykinesia
- Akinetic mutism: inability to move or speak
- Motor impersistence: inability to sustain simple voluntary acts (e.g., tongue protrusion)
- Dysarthria and dysphagia
- Cognitive decline, psychiatric symptoms, and behavioral changes (these symptoms may mimic substance use)
- Dementia (particularly executive dysfunction)
- Major depressive disorder (possibly including suicidal tendencies)
- Schizophrenia-like psychosis (∼ 10% of cases)
- Paranoid delusions (most common), delusions of infidelity
- Auditory hallucinations
- Aggression
- Movement dysfunction
Tip
Chorea characterizes the early stages of the disease while hypokinetic/akinetic symptoms may dominate later on. Dementia, depression, and behavioral disorders are common in advanced stages.
Diagnostics
- Genetic testing (e.g. polymerase chain reaction)
- CT/MRI: atrophy of the striatum, most pronounced in the caudate nucleus with consequent enlargement of ventricles (ex vacuo ventriculomegaly) t

Differential diagnostics
Ballismus
Tip
- Differentiation: Ballismus vs. Huntington
- Movement Character:
- Ballismus: Violent, flailing, large amplitude. Proximal muscles.
- Huntington: Chorea (jerky, “dance-like”), low amplitude. Distal muscles/face.
- Lesion Location:
- Ballismus: Subthalamic Nucleus.
- Huntington: Caudate Nucleus.
- Onset:
- Ballismus: Acute (usually 2° to Lacunar Stroke).
- Huntington: Gradual (Neurodegenerative, age 20–50).
- Definition & Clinical Presentation
- Characterized by large-amplitude, violent, flailing movements of the limbs.

- Involves proximal musculature (shoulders, hips), distinguishing it from chorea (distal, dance-like).
- Most commonly presents unilaterally as Hemiballismus.
- Characterized by large-amplitude, violent, flailing movements of the limbs.
- Lesion Localization
- Contralateral Subthalamic Nucleus (STN). t
- Etiology
- Most common: Lacunar Stroke (ischemic infarction) usually secondary to long-standing hypertension.
- Other causes: Non-ketotic hyperglycemia (HHNK), neoplasms, HIV, SLE.
- Pathophysiology (Basal Ganglia Circuitry)
- Normal Circuit: STN stimulates the Globus Pallidus internus (GPi) GPi inhibits the Thalamus.
- Lesion Circuit: Damage to STN excitation of GPi inhibition of Thalamus.
- Result: Unopposed thalamic output to the motor cortex Hyperkinetic movement.
- Treatment
- Dopamine Antagonists: High-potency antipsychotics (e.g., Haloperidol) allow for movement suppression.
- Depleting agents: Tetrabenazine.
- Prognosis: Often self-limited; may resolve spontaneously over weeks/months.