Epidemiology
Etiology
Pathophysiology
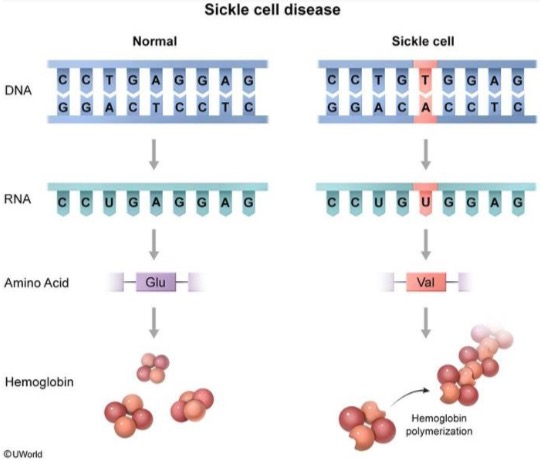
- Point mutation in the β-globin gene (chromosome 11) → glutamic acid replaced with valine (single amino acid substitution) → 2 α-globin and 2 mutated β-globin subunits create pathological hemoglobin S (HbS).
- Heterozygotes (HbAS): carry one sickle allele and one other (usually normal) → sickle cell trait
- Homozygotes (HbSS): carry two sickle alleles → sickle cell anemia
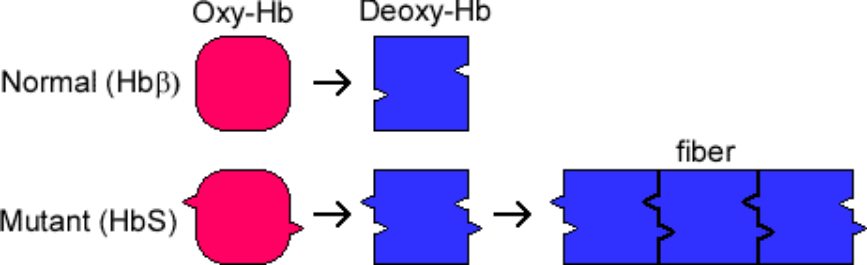
- HbS polymerizes when deoxygenated, causing deformation of erythrocytes (“sickling”). t This can be triggered by any event associated with reduced oxygen tension.
- Formation of a hydrophobic pocket on the beta globin surface that interacts with a complementary nonpolar residue on another hemoglobin molecule. The hydrophobic interaction causes polymerization of HbS molecules and subsequent erythrocyte sickling, leading to membrane damage and permanent distortion of red blood cells.
- Hypoxia (e.g., at high altitudes)
- In homozygotes, up to 100% of the hemoglobin molecules are affected, leading to sickle cell formation under minimally decreased oxygen tension.
- In heterozygotes, sickling only occurs due to severe reduction in oxygen tension.
- Infections
- Dehydration
- Acidosis
- Sudden changes in temperature
- Stress
- Formation of a hydrophobic pocket on the beta globin surface that interacts with a complementary nonpolar residue on another hemoglobin molecule. The hydrophobic interaction causes polymerization of HbS molecules and subsequent erythrocyte sickling, leading to membrane damage and permanent distortion of red blood cells.
- Sickle cells lack elasticity and adhere to vascular endothelium, which disrupts microcirculation and causes vascular occlusion and subsequent tissue infarction.
- Hemolysis and the subsequent increased turnover of erythrocytes may increase the demand for folate, causing folate deficiency.
- Can present as macrocytic anemia
- Extravascular hemolysis and intravascular hemolysis are common and result in anemia.
Clinical features
Typically manifests after 3–6 months of age as the production of HbF decreases and HbS levels increase
Acute manifestations
- Early Infancy: Patients are often asymptomatic for the first 6 months due to high levels of fetal hemoglobin (HbF), which inhibits sickling.
- Vaso-occlusive Crisis (VOC): The most common manifestation, causing acute, severe pain in the bones (especially long bones), joints, abdomen, and chest.
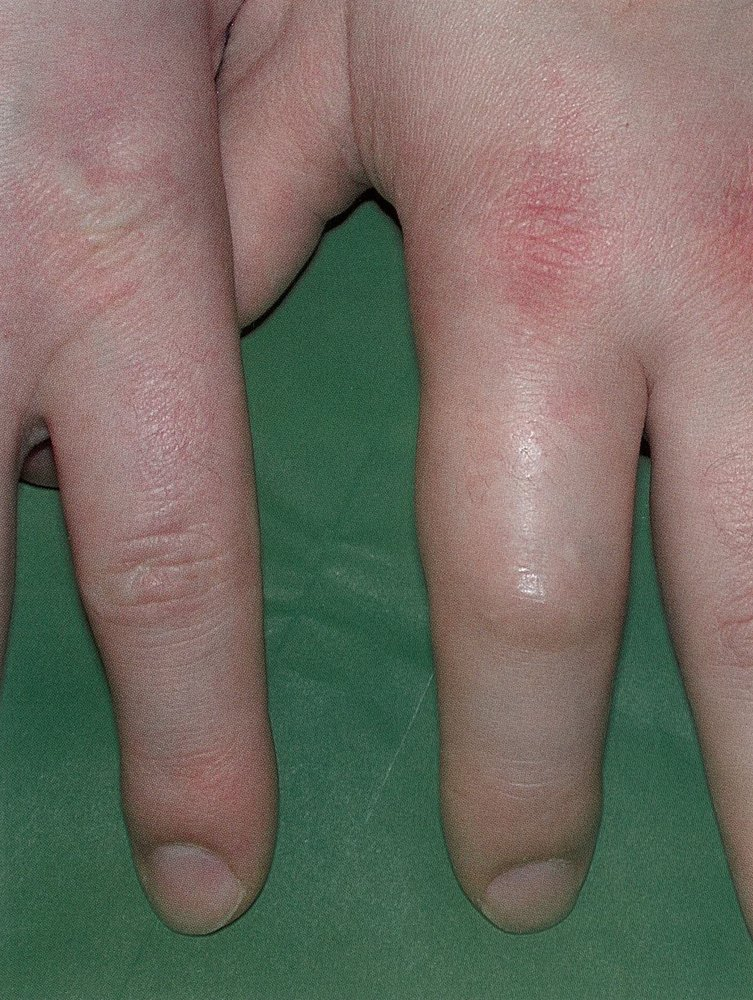
- Dactylitis: Often the first sign in children (6 months - 4 years), presenting as painful swelling of the hands and feet (“hand-foot syndrome”).
- Anemia: Chronic hemolytic anemia leads to fatigue, pallor, and jaundice.
- Splenic Sequestration Crisis: A life-threatening complication in young children where a large volume of blood pools in the spleen, causing severe anemia, hypovolemia, and a rapidly enlarging spleen.
Diagnostics
- Normal RBC indices (e.g. MCV)
- Not all red blood cells are sickled
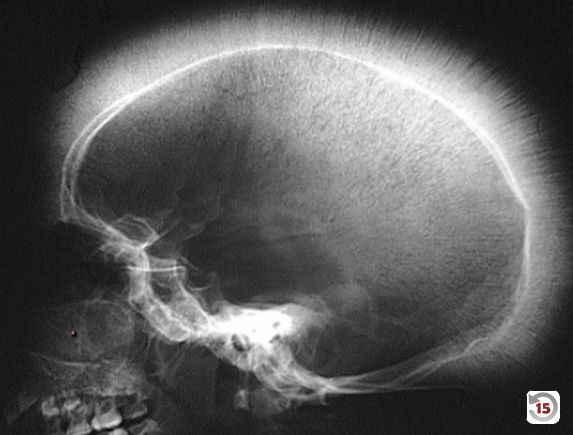
- Skull x-ray: Hair-on-end (also known as “crew cut”) sign, caused by excessive expansion of the bone marrow to compensate for the destruction of red blood cells
Treatment
Overview
Infants and children
- Antibiotic prophylaxis against invasive pneumococcal disease until 5 years of age
- Hydroxyurea therapy regardless of clinical severity to minimize disease-related complications (First-line)
- Annual transcranial doppler to screen for stroke risk from 2 months till 16 years of age
Hydroxyurea therapy
Indications
- All infants > 9 months, children, and adolescents with sickle cell anemia regardless of symptom severity
- Adults with any of the following:
- Frequent (≥ 3 episodes/year) acute pain episodes or other vasoocclusive events
- Severe symptomatic anemia
- History of severe and/or recurrent acute chest syndrome
Mechanism of action
Stimulation of erythropoiesis and increased fetal hemoglobin
Complications
Splenic sequestration
- Pathophysiology: splenic vasoocclusion and entrapment and pooling of large amounts of blood in the spleen
- Clinical presentation
- Most commonly affects children < 5 years of age
- Acute left upper quadrant pain, splenomegaly
- Hypotension and/or hypovolemic shock, especially in infants
- Due to the trapping of blood in the spleen
- Symptoms of anemia (e.g., pallor, fatigue)
- Functional asplenia: Increased risk of infection with encapsulated bacteria (Streptococcus pneumoniae (most common), Neisseria meningitis, Haemophilus influenzae type b
- Supportive findings
- Anemia (Hb drop of ≥ 2 g/dL) with reticulocytosis
- Thrombocytopenia
- Acute management
- Immediate IV fluid resuscitation for hypovolemia
- Simple RBC transfusion in consultation with a sickle cell expert (avoid raising hemoglobin > 8 g/dL)
Infections
- Infections: Functional asplenia from repeated splenic infarctions leads to increased susceptibility to encapsulated organisms (S. pneumoniae, H. influenzae type B, N. meningitidis). Sepsis is a major cause of death in children.