Etiology
CF is a hereditary autosomal recessive t disorder caused by defective CFTR (cystic fibrosis transmembrane conductance regulator) protein due to mutation in the CFTR gene located on the long arm of chromosome 7.
Pathophysiology
- Mutated CFTR gene → misfolded protein (impaired post-translational) → degradation of the defective protein in the rough endoplasmic reticulum (rER) → absence of ATP-gated chloride channel on the cell surface of epithelial cells throughout the body (e.g., intestinal and respiratory epithelia, Hair follicles and glands, exocrine pancreas, exocrine glands of reproductive organs)
- In Hair follicles and glands
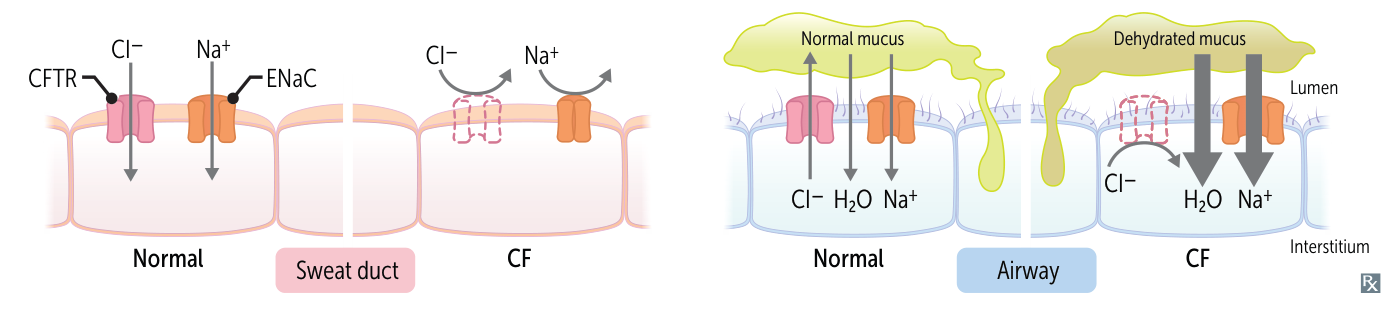
- Defective ATP-gated chloride channel → inability to reabsorb Cl- from the lumen of the Hair follicles and glands → reduced reabsorption of Na+ and H2O → excessive loss of salt and elevated levels of NaCl in sweat
- Because Cl- attracts Na+
- In all other exocrine glands (e.g., in the GI tract or lungs)
- Defective ATP-gated chloride channel → inability to transport intracellular Cl- across the cell membrane → reduced secretion of Cl- and H2O → accumulation of intracellular Cl- → ↑ Na+ reabsorption (via ENaC) → ↑ H2O reabsorption → formation of hyperviscous mucus → accumulation of secretions and blockage of small passages of affected organs → chronic inflammation and remodeling → organ damage
- ↑ Na+ reabsorption → transepithelial potential difference between interstitial fluid and the epithelial surface increases (i.e., negative charge increases; e.g., from normal -13 mv to abnormal -25 mv)
Clinical features

GI
- Meconium ileus (in newborns)
- Failure to pass the first stool in neonates (meconium usually passes within the first 24–48 hours after birth)
- Etiology: Cystic fibrosis is the cause in > 90% of cases.
- Clinical findings: signs of a distal small bowel obstruction (thick meconium plugs the distal ileum)
- Bilious vomiting
- Abdominal distention
- No passing of meconium or stool
- Failure to thrive (due to malabsorption)
- Pancreatic disease (obstruction of pancreatic ducts)
- Autopsy revealed pancreas develops cysts and became scarred, that’s how it’s named cystic fibrosis
- Pancreatitis
- Exocrine pancreatic insufficiency
- Foul-smelling steatorrhea (fatty stools) may occur.
- Malabsorption
- Abdominal distention
- Diarrhea
- Hypoproteinemia
- Deficiency of fat-soluble vitamins
- CF-related diabetes mellitus (CFRD)
- Liver and bile duct abnormalities
- Biliary cirrhosis with portal hypertension, jaundice, and/or esophageal varices
- Intestinal obstruction: abdominal distention, pain, and a palpable mass
- Rectal prolapse (rare)
Tip
In almost all cases of meconium ileus, cystic fibrosis is the underlying disease.
Respiratory
- COPD with bronchiectasis
- Chronic sinusitis: nasal polyps may eventually develop
- Recurrent or chronic productive cough and pulmonary infections
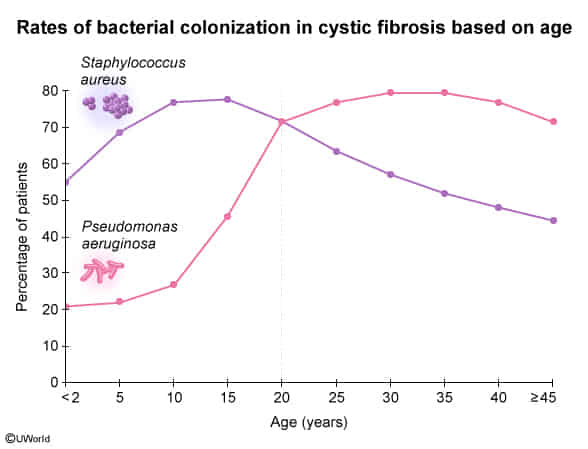
- S. aureus is the most common cause of recurrent pulmonary infection in infancy and childhood.
- P. aeruginosa is the most common cause of recurrent pulmonary infections in adulthood.
- Other High-Yield Pathogens:
- Methicillin-resistant S. aureus (MRSA).
- Burkholderia cepacia complex: Associated with accelerated decline in lung function and increased mortality. t
- Nontuberculous mycobacteria (NTM).
- Hemoptysis (Recurrent infections lead to increased vulnerability of the lung tissue)
- Signs of chronic respiratory insufficiency: digital clubbing
bronchiectasis (irreversible) → progressive respiratory failure → death
The main determinant of life expectancy is the severity of pulmonary disease: chronic respiratory infections and mucus plugging →
Hair follicles and glands
- Particularly salty sweat
- Possible electrolyte wasting
Genital
- Men: usually infertile
- Obstructive azoospermia is common, spermatogenesis may be intact
- The vas deferens may be absent.
- CF causes the vas deferens to atrophy early in embryologic development.
- Women: reduced fertility
- Thick cervical mucus is harder for the sperm to travel through
Diagnostics
Confirmatory tests
- Sweat test (quantitative pilocarpine iontophoresis)
- Indications: preferred initial test in all patients with suspected CF
Newborn screening
Newborn screening (NBS) for CF is essential for early detection and treatment, which can improve health outcomes. In many countries, including the US, it is mandatory to screen for CF.
- Sample collection: newborn blood spot test, performed via heel prick blood sampling in the first 24 and 48 hours of life
- Screening tests
- Immunoreactive trypsinogen (IRT) : initial screening test
- IRT levels are thought to become elevated due to mucus plugs, which cause pancreatic duct obstruction, preventing the conversion of trypsinogen to trypsin. This can occur in neonates with or without pancreatic insufficiency.
- An immunofluorescence assay that measures levels of IRT
- Normal IRT levels: CF unlikely
- Elevated IRT levels: CF possible; additional screening tests required
- CFTR mutation testing (DNA testing): second-tier screening test
- Immunoreactive trypsinogen (IRT) : initial screening test