| Aspect | Progressive Muscular Dystrophies | Myotonic Syndromes |
|---|---|---|
| Definition | A group of genetic disorders characterized by progressive muscle weakness and wasting. | Disorders characterized by muscle weakness along with myotonia (difficulty relaxing muscles after contraction). |
| Main Types | Duchenne, Becker | Type 1 (DM1) and Type 2 (DM2) |
| Inheritance | Often X-linked (e.g., Duchenne/Becker) or autosomal (e.g., Limb-Girdle). | Autosomal dominant. Trinucleotide repeat expansion diseases |
| Onset | Usually in childhood or adolescence. | Typically adolescence or adulthood. |
| Primary Symptoms | Muscle weakness, wasting, contractures, scoliosis, cardiac complications. | Muscle weakness, myotonia, cataracts, cardiac arrhythmias, endocrine issues. |
| Progression | Gradual; severity and speed depend on the subtype. | Gradual; multisystem involvement often worsens with age. |
| Pathophysiology | Defects in proteins like dystrophin (Duchenne/Becker) or other sarcolemmal proteins. | Expanded CTG (DM1) or CCTG (DM2) repeats causing RNA toxicity and protein misregulation. |
| Diagnosis | Genetic testing, muscle biopsy, creatine kinase (CK) levels, EMG. | Genetic testing, EMG (shows myotonic discharges). |
| Treatment | Symptomatic: physical therapy, steroids (e.g., Duchenne), assistive devices. | Symptomatic: medications for myotonia (e.g., mexiletine), physical therapy. |
| Cardiac Involvement | Common, especially in Duchenne and Becker (cardiomyopathy). | Common, often conduction abnormalities or arrhythmias. |
| Respiratory Complications | Can develop in advanced stages due to diaphragm involvement. | Can occur due to muscle weakness or central control issues. |
| Cognitive Impact | Rare | Can include mild cognitive and behavioral changes. |
| Prognosis | Varies by type; some forms (e.g., Duchenne) are life-limiting. | Varies by severity; generally better than PMD but depends on systemic involvement. |
Epidemiology
- Sex: only male individuals affected in DMD and BMD
- Age of onset
- DMD: 2–5 years
- BMD: adolescence or early adulthood, usually > 15 years
Etiology
- Inheritance pattern (DMD and BMD): X-linked recessive
- Chromosomal mutations affecting the dystrophin gene on the short arm of the X chromosome (Xp21)
- DMD: frameshift deletion or nonsense mutation → shortened or absent dystrophin protein
- BMD: in-frame deletion → partially functional dystrophin protein
- In about two-thirds of DMD or BMD cases, deleted segments are as large as one or more exons.
Pathophysiology
- Dystrophin protein: anchors the cytoskeleton of skeletal and cardiac muscle cells to the extracellular matrix by connecting cytoskeletal actin filaments to membrane-bound α- and β-dystroglycan, which are connected to extracellular laminin
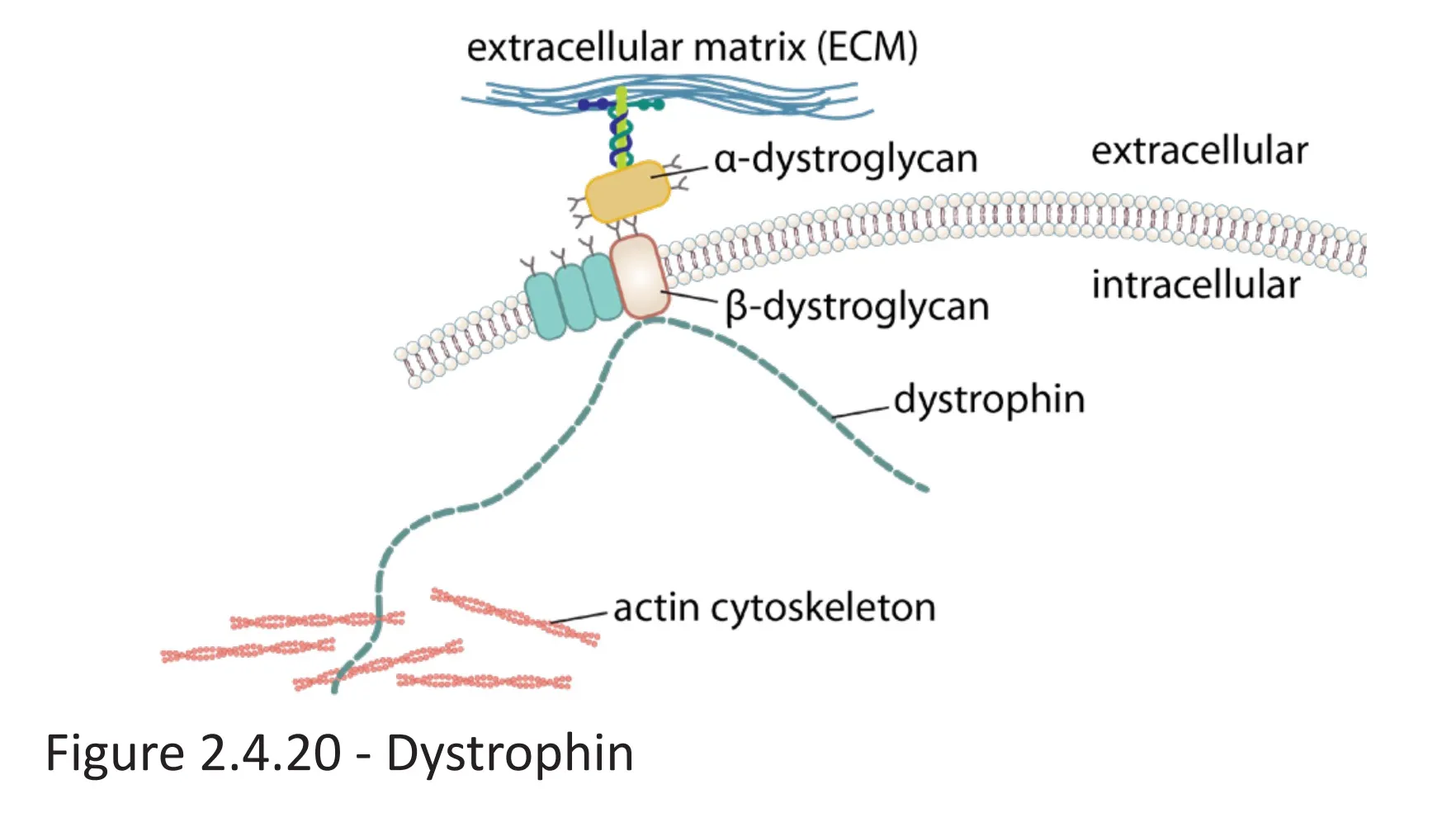
- Dystrophin gene: largest known protein-coding gene in the human DNA
- Because of its size, the dystrophin gene is at increased risk for spontaneous mutations.
- Mutations affecting the dystrophin gene→ alterations of dystrophin protein structure → partial (BMD) or almost complete (DMD) impairment of protein function → disturbance of numerous cellular signaling pathways → necrosis of affected muscle cells → replacement with connective tissue and fatty tissue → affected muscles are weak even though they appear larger (“pseudohypertrophy”)
- Errors in splicing in the DMD gene can also cause DMD.
Clinical features
Duchenne muscular dystrophy (DMD)
- Progressive muscle paresis and atrophy
- Starts in the proximal lower limbs (pelvic girdle)
- Extends to the upper body and distal limbs as the disease progresses
- Weak reflexes
- Waddling gait (i.e., Duchenne limp) with bilateral Trendelenburg sign
- Gower maneuver
- The individual arrives at a standing position by supporting themselves on their thighs and then using the hands to “walk up” the body until they are upright.
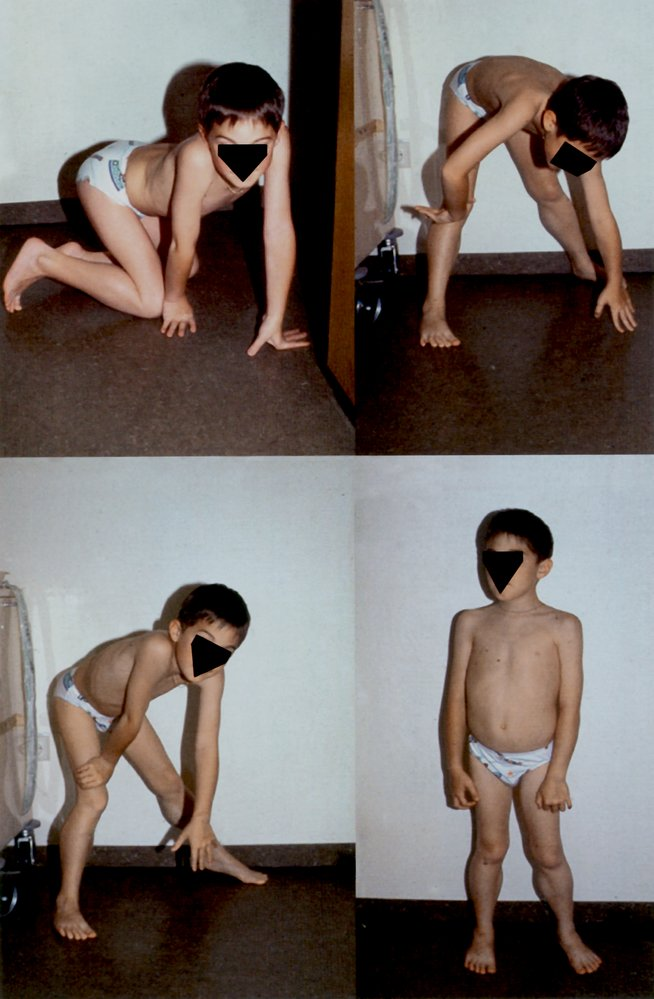
- Classic sign of DMD, but also occurs in inflammatory myopathies (e.g., dermatomyositis, polymyositis) and other muscular dystrophies (e.g., BMD)
- The individual arrives at a standing position by supporting themselves on their thighs and then using the hands to “walk up” the body until they are upright.
- Calf pseudohypertrophy
- Mutations affecting the dystrophin gene→ alterations of dystrophin protein structure → partial (BMD) or almost complete (DMD) impairment of protein function → disturbance of numerous cellular signaling pathways → necrosis of affected muscle cells → replacement with connective tissue and fatty tissue → affected muscles are weak even though they appear larger (“pseudohypertrophy”)
Becker muscular dystrophy (BMD)
- Symptoms similar to those of DMD, but less severe
- Slower progression (patients often remain ambulatory into adult life)
Mnemonic
Becker is better