- Amyloid: insoluble protein or protein fragments
- Amyloidosis: extracellular aggregation and subsequent deposition of amyloid in various organs


AL amyloidosis (primary amyloidosis)
Most common form of amyloidosis
Etiology
Low-level expansion of a plasma cell dyscrasia (e.g., multiple myeloma, Waldenstrom macroglobulinemia)
Pathophysiology
Increased production of the light chains of immunoglobulins → deposition of amyloid light chain protein (AL protein)
Clinical features
- Heart: restrictive cardiomyopathy, HFpEF, atrioventricular block
- Kidney: nephrotic syndrome, type II renal tubular acidosis, nephrogenic diabetes insipidus
- Tongue: macroglossia → obstructive sleep apnea
- Autonomic nervous system: autonomic neuropathy
- Musculoskeletal system
- Carpal tunnel syndrome (may be bilateral)
Diagnostics
Treatment
AA amyloidosis (secondary amyloidosis)
Etiology
AA amyloidosis is secondary to a chronic disease, such as:
- Chronic inflammatory conditions (e.g., IBD, rheumatoid arthritis, SLE, vasculitis, familial Mediterranean fever)
- Currently, rheumatic diseases such as rheumatoid arthritis (RA), ankylosing spondylitis (AS), psoriatic arthritis, and juvenile idiopathic arthritis are the most frequent causes (70%) of AA amyloidosis.
- Chronic infectious diseases (e.g., tuberculosis, bronchiectasis, leprosy, osteomyelitis)
Pathophysiology
Chronic inflammatory process → ↑ production of acute phase reactant SAA (serum amyloid-associated protein) → deposition of AA (amyloid-associated) protein in various organs
Clinical features
- Kidney: nephrotic syndrome
- Liver and spleen: hepatomegaly, splenomegaly
Tip
The main feature of AA amyloidosis at diagnosis is renal dysfunction (e.g., CKD, nephrotic syndrome). Cardiac involvement is rare.
Diagnostics
Tip
Amyloidosis should always be considered in patients who present with kidney, liver, or GI involvement in the setting of chronic inflammatory and/or infectious disease.
Treatment
ATTRmt amyloidosis
- Autosomal dominant disease (most common) that is due to mutated transthyretin (ATTR)
- Familial amyloid cardiomyopathy (FAC)
- Common in African Americans (3% of African Americans carry the mutant allele for FAC)
- Deposition in ventricular endomyocardium → restrictive cardiomyopathy
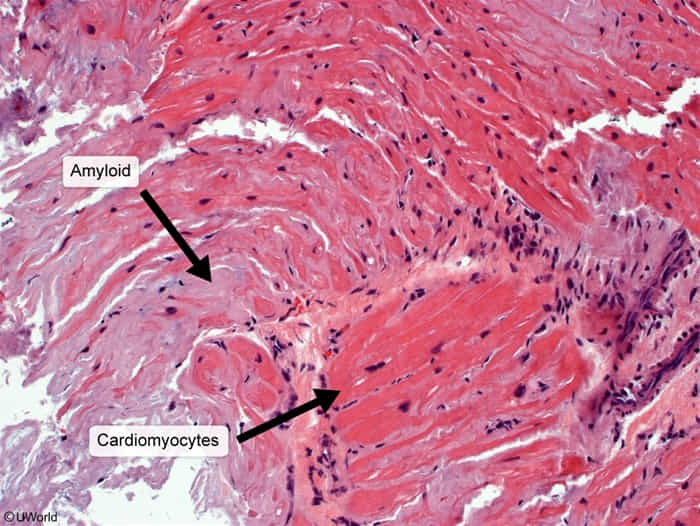
- Atrial deposition → arrhythmia