- Impaired copper excretion causes copper to accumulate in the body.
- Early-stage Wilson disease is characterized by the presence of copper deposits in the liver.
- As the disease progresses, copper accumulates in other organs as well, most importantly in the brain and cornea.
Epidemiology
Etiology
Pathophysiology
Tip
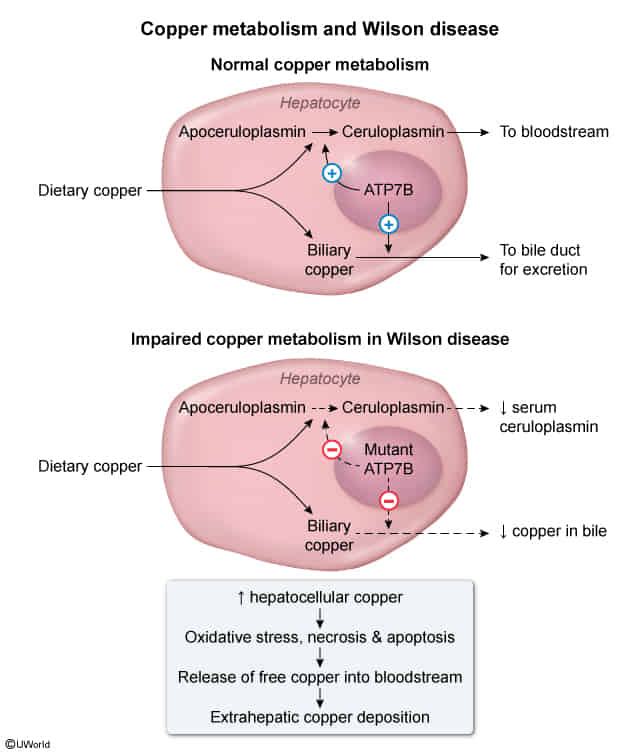
- Apoceruloplasmin
- Copper-free precursor synthesized in liver.
- Unstable half-life; degrades rapidly if not loaded with copper.
- Ceruloplasmin
- Copper-bound form (loaded by ATP7B in Golgi).
- Stable half-life; carries 95% of circulating copper.
- Ferroxidase activity (links Cu metabolism to Fe metabolism).
- Autosomal Recessive mutation of ATP7B gene on Chromosome 13.
- Defect in hepatocyte copper-transporting ATPase (ATP7B protein).
- Primary Defects:
- Inability to excrete excess copper into bile (main route of excretion).
- Failure to incorporate copper into apoceruloplasmin to form functional ceruloplasmin.
- Consequences:
- ↓ Serum ceruloplasmin (due to shorter half-life of apoceruloplasmin).
- ↑ Free (unbound) serum copper.
- Accumulation of toxic copper levels in tissues via Fenton reaction (oxidative damage).
- Tissue Deposition Sequence:
- Liver: Accumulates first → hepatocyte necrosis → micronodular cirrhosis.
- Brain: Deposits in Basal Ganglia (specifically Putamen and Globus Pallidus) → parkinsonism, dysarthria, tremor.
- Cornea: Deposits in Descemet membrane → Kayser-Fleischer rings.
- Kidneys: Proximal tubule damage → Fanconi syndrome.
Tip
Don’t mess up with Hemochromatosis
Clinical features
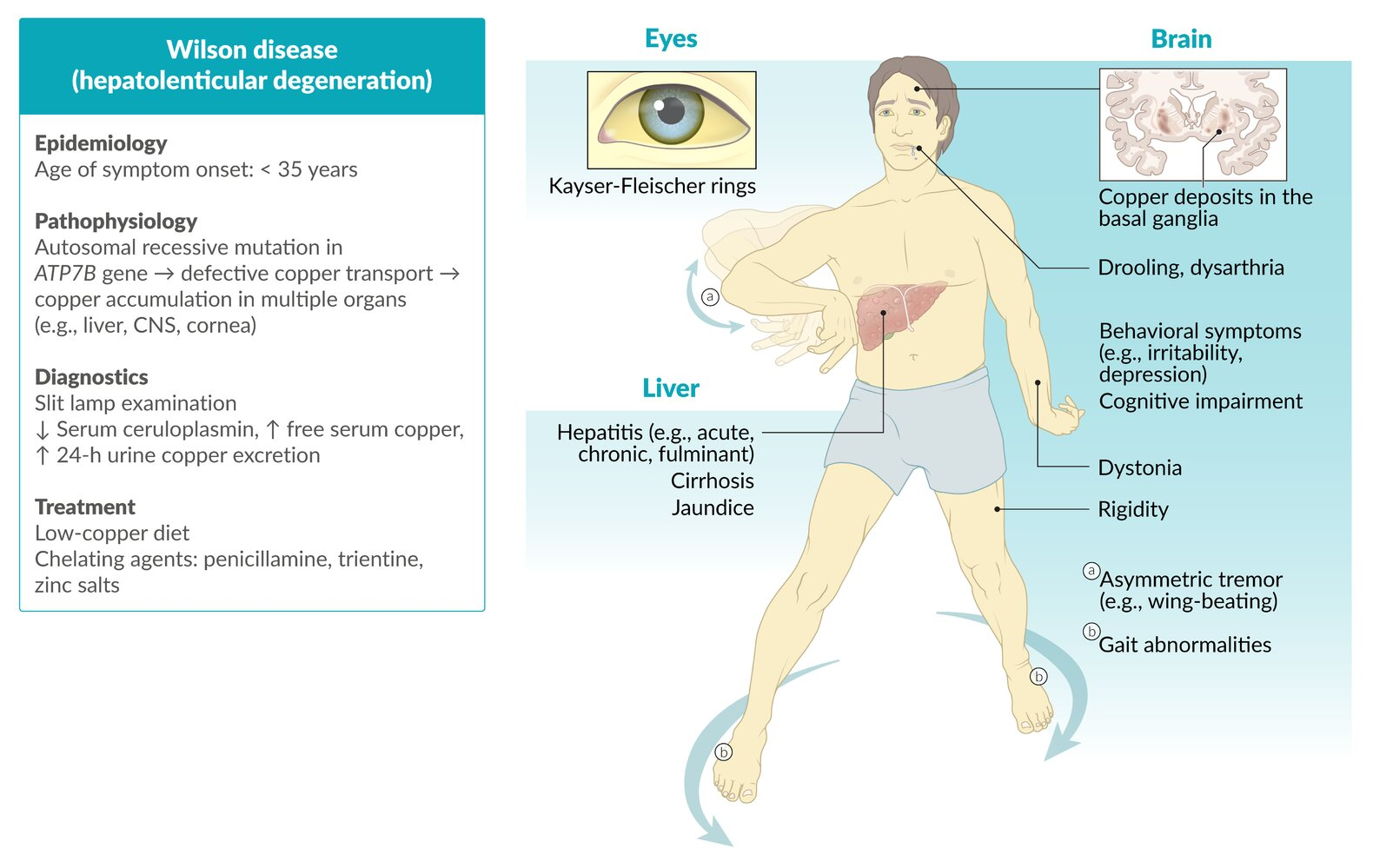
- Neurological
- Cerebellar symptoms, e.g.:
- Dysarthria (most common)
- Extrapyramidal symptoms, e.g.:
- Dystonia
- Parkinsonism
- Tremor (usually asymmetric, affecting the hands), which may be any combination of:
- Resting tremor
- Intention tremor
- Wing-beating tremor: a low frequency, high amplitude tremor that is most prominent when the arms are outstretched anteriorly or laterally
- Drooling (caused by oropharyngeal dysphagia)
- Cognitive impairment
- Cerebellar symptoms, e.g.:
Wilson disease vs hemochromatosis
- Wilson disease has neurologic symptoms but hemochromatosis doesn’t have
- Think about Wilson from Don’t Starve, the mad scientist

Diagnostics
Treatment
General principles
- Encourage a low-copper diet (e.g., avoidance of organ meats, shellfish, nuts, chocolate, copper-containing dietary supplements).
- Refer patients with refractory decompensated cirrhosis or acute liver failure for liver transplantation.
Pharmacological therapy
- First line: chelating agents, e.g., penicillamine (preferred) or trientine
- Chelating agents facilitate renal excretion of copper by forming water-soluble compounds.
- Adverse effect: Membranous nephropathy
- Maintenance therapy: reduced-dose zinc salts or a chelating agent