Epidemiology
Etiology
Primary adrenal insufficiency (Addison disease)
- Destruction of the adrenal cortex leading to ↓ cortisol AND ↓ aldosterone.
- Most common cause in developed countries: Autoimmune adrenalitis.
- Pathology
- Gross: Atrophic, shrunken glands.
- Micro: Lymphocytic infiltration t (sparse/absent cortex).
- Pathology
- Other causes: Infections (TB, HIV, fungal), bilateral adrenal hemorrhage (Waterhouse-Friderichsen syndrome in meningococcemia), metastasis.
Secondary & tertiary adrenal insufficiency
- Secondary: Decreased ACTH secretion from the pituitary (e.g., pituitary tumors, Sheehan syndrome).
- Tertiary: Decreased CRH from the hypothalamus. Most common cause: Chronic exogenous steroid use followed by abrupt cessation.
- Leads to ↓ cortisol ONLY. Aldosterone is preserved because it is primarily regulated by the Renin-Angiotensin-Aldosterone System (RAAS), not ACTH.
| Feature | Primary AI | Secondary/Tertiary AI |
|---|---|---|
| Defect | Adrenal Gland Failure | ↓ ACTH / CRH (Pituitary/Hypothalamus) |
| ACTH | ↑ High | ↓ Low |
| Aldosterone | ↓ Low | Normal (RAAS intact) |
| Key Signs | Hyperpigmentation & Hyperkalemia | NO hyperpigmentation or hyperkalemia |
| Treatment | Cortisol + Fludrocortisone | Cortisol ONLY |
Pathophysiology
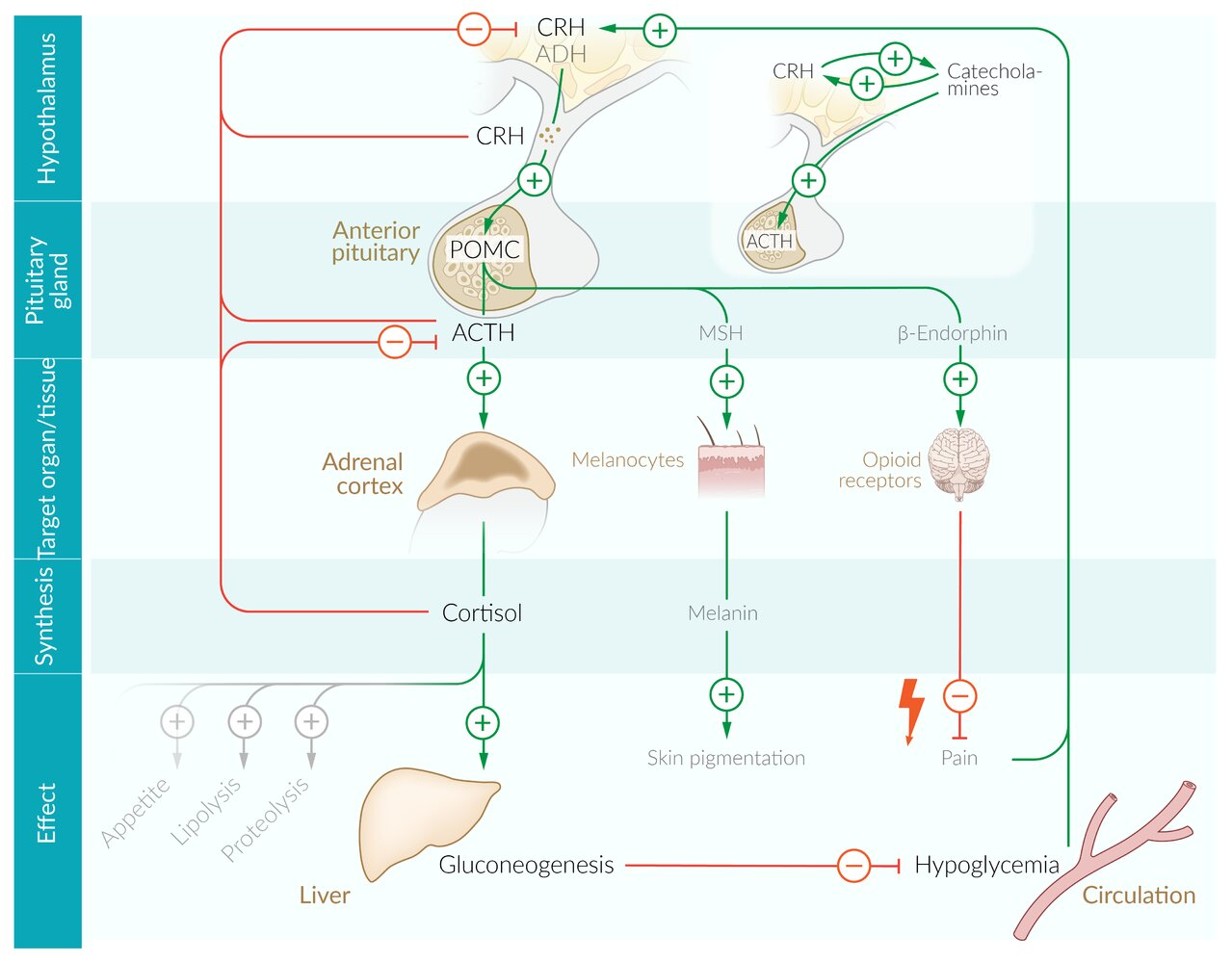
Primary adrenal insufficiency (Addison disease)
Damage to the adrenal gland leads to the deficiency in all three hormones produced by the adrenal cortex: androgen, cortisol, and aldosterone.
- Hypoandrogenism
- Loss of libido
- Impaired spermatogenesis (in men)
- Hypocortisolism leads to:
- ↑ ACTH → ↑ production of POMC (in order to increase ACTH production) → ↑ melanocyte-stimulating hormone (MSH) → hyperpigmentation of the skin (bronze skin)
- ↑ ADH level → retention of free water → dilutional hyponatremia
- ↓ Expression of enzymes involved in gluconeogenesis → ↓ rate of gluconeogenesis → hypoglycemia
- Lack of potentiation of catecholamines action → hypotension
- Hypoaldosteronism → hypotension (hypotonic hyponatremia and volume contraction), hyperkalemia, metabolic acidosis
Clinical features
- Shared Features (due to ↓ Cortisol):
- Constitutional: Fatigue, weakness, weight loss, anorexia.
- GI: Nausea, vomiting, abdominal pain, diarrhea.
- Orthostatic hypotension and dizziness.
- Hypoglycemia, especially in children.
- Permissive action
- Reverse of 诱发三高和溃疡
- Features specific to Primary AI (due to ↓ Aldosterone & ↑ ACTH):
- Hyperpigmentation: Caused by ↑ ACTH which also stimulates melanocytes (MSH). Visible on skin creases, scars, buccal mucosa.
- In secondary AI, ACTH is low; therefore, no hyperpigmentation would be seen. t
- Salt craving.
- Hyperkalemia and hyponatremia (due to aldosterone deficiency).
- More severe hypotension/volume depletion.
- Hyperpigmentation: Caused by ↑ ACTH which also stimulates melanocytes (MSH). Visible on skin creases, scars, buccal mucosa.
- Acute Adrenal Crisis:
- Life-threatening medical emergency.
- Precipitated by stress (infection, trauma, surgery) in a patient with underlying insufficiency. t
- Sx: Profound hypotension (shock refractory to fluids), fever, confusion, vomiting.

Diagnostics
- Initial Labs:
- Hyponatremia and hyperkalemia (hallmarks of primary disease due to low aldosterone).
- Hypoglycemia.
- Eosinophilia may also be present.
- Glucocorticoids are potent anti-inflammatory agents. One of their key immune-modulating effects is to suppress eosinophils.
- Morning cortisol level: A low level is suggestive of adrenal insufficiency.
- Confirmatory Test:
- The ACTH stimulation test (Cosyntropin test) is the gold standard for diagnosis.
- A baseline cortisol level is drawn, followed by an IV administration of synthetic ACTH (cosyntropin). Cortisol levels are measured again at 30 and 60 minutes.
- Normal response: A significant rise in cortisol levels (e.g., to >18-20 mcg/dL).
- Adrenal insufficiency: An insufficient or absent rise in cortisol levels.
- Differentiating Primary vs. Secondary:
- Plasma ACTH level:
- High in primary adrenal insufficiency (due to loss of negative feedback).
- Low or inappropriately normal in secondary/tertiary adrenal insufficiency.
- Overnight metyrapone stimulation test t
- Mechanism/Physiology
- Metyrapone inhibits 11--hydroxylase in the adrenal steroid synthesis pathway.
- Blocking this enzyme prevents the conversion of 11-deoxycortisol to cortisol.
- Resulting cortisol leads to loss of negative feedback on the pituitary and hypothalamus.
- Normal physiologic response: Compensatory surge in ACTH secretion adrenal stimulation accumulation of the precursor 11-deoxycortisol (which is measurable in urine as 17-hydroxycorticosteroids).
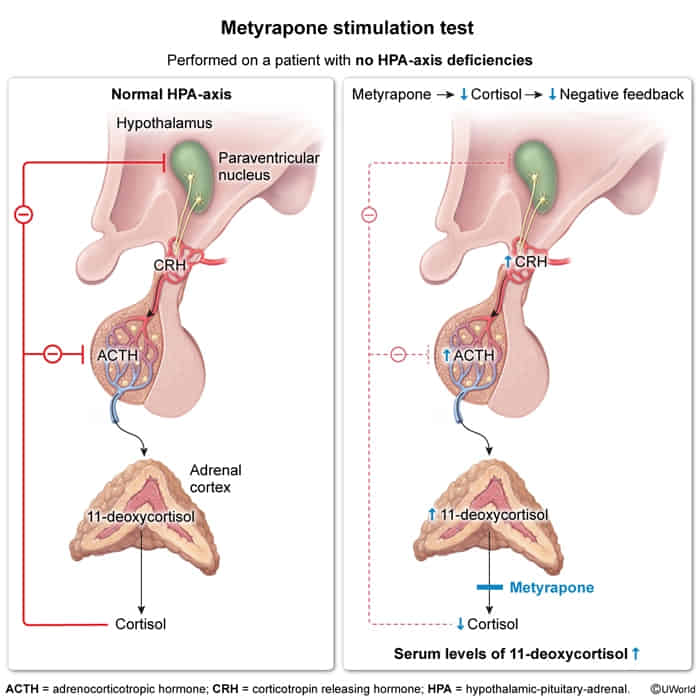
- Normal Result: ACTH 11-deoxycortisol (precursor).
- Primary AI (Addison): ACTH (baseline high), 11-deoxycortisol (adrenal cannot respond).
- Secondary AI (Pituitary): ACTH, 11-deoxycortisol (pituitary cannot release ACTH).
- Mechanism/Physiology
- Plasma ACTH level:
Treatment
Steroid replacement
- Glucocorticoids
- Hydrocortisone
- Cortisone acetate
- Prednisolone
- Mineralocorticoids
- Agent: fludrocortisone (a synthetic mineralocorticoid with mostly mineralocorticoid and limited glucocorticoid effects)
Adrenal crisis
Adrenal crisis is an acute, severe glucocorticoid deficiency that requires immediate emergency treatment.
Precipitating factors for adrenal crisis
- Stress in patients with underlying adrenal insufficiency e.g.:
- Gastrointestinal illness (most common)
- Other infections
- Perioperative period
- Sudden discontinuation of glucocorticoids after prolonged glucocorticoid therapy
- Bilateral adrenal hemorrhage or infarction (e.g., Waterhouse-Friderichsen syndrome)
- Pituitary apoplexy
Tip
In order to prevent the development of secondary and tertiary adrenal insufficiency, prolonged steroid therapy should be tapered slowly rather than stopped abruptly.
Signs and symptoms
- Hypotension, shock
- Impaired consciousness, coma
- Fever
- Vomiting, diarrhea
- Severe abdominal pain (which can resemble peritonitis)
Autoimmune polyglandular syndromes
- Definition: a set of conditions characterized by autoimmune disease that causes multiple endocrine deficiencies, which affect the hormone-producing (endocrine) glands
- Types
- Type 1: (APS-1, Whitaker syndrome, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, or APECED)
- Caused by a mutation in the autoimmune regulator gene (AIRE)
- Facilitates the elimination of self-reactive T cells.
- Associated endocrine deficiencies (two or more of the following should be present)
- Most commonly
- Primary adrenal insufficiency
- Hypoparathyroidism
- Chronic mucocutaneous candidiasis
- Ectodermal dystrophy of skin, nails, and dental enamel
- Most commonly
- Caused by a mutation in the autoimmune regulator gene (AIRE)
- Type 2 (APS-2, Schmidt syndrome): defined by the occurrence of primary adrenal insufficiency with thyroid autoimmune disease and/or type 1 diabetes mellitus
- Main manifestation: primary adrenal insufficiency
- Associated endocrine deficiencies (one or more of the following may be present)
- Most commonly
- Thyroid autoimmune disease (e.g., Hashimoto thyroiditis)
- Type 1 diabetes mellitus
- Most commonly
- Associated endocrine deficiencies (one or more of the following may be present)
- Main manifestation: primary adrenal insufficiency
- Type 1: (APS-1, Whitaker syndrome, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, or APECED)
- High-Yield Management Pearl
- When treating a patient with both adrenal insufficiency and hypothyroidism, always initiate corticosteroids BEFORE levothyroxine.
- Rationale: Thyroid hormone increases metabolic clearance of cortisol; starting levothyroxine first in an adrenal-insufficient patient can precipitate an adrenal crisis.