Epidemiology
- Beta thalassemia: most commonly seen in people of Mediterranean descent
- Alpha thalassemia: most commonly seen in people of Asian and African descent
Mnemonic
Alpha thalassemia is common in Asia and Africa.
Etiology
- Beta thalassemia: usually due to point mutations in promoter sequences or splicing sites
- β-globin locus - short arm of chromosome 11
- Alpha thalassemia: usually due to deletion of at least one out of the four existing alleles
- The α-globin gene cluster is located on chromosome 16
Pathophysiology
- Inefficient erythropoiesis → anemia
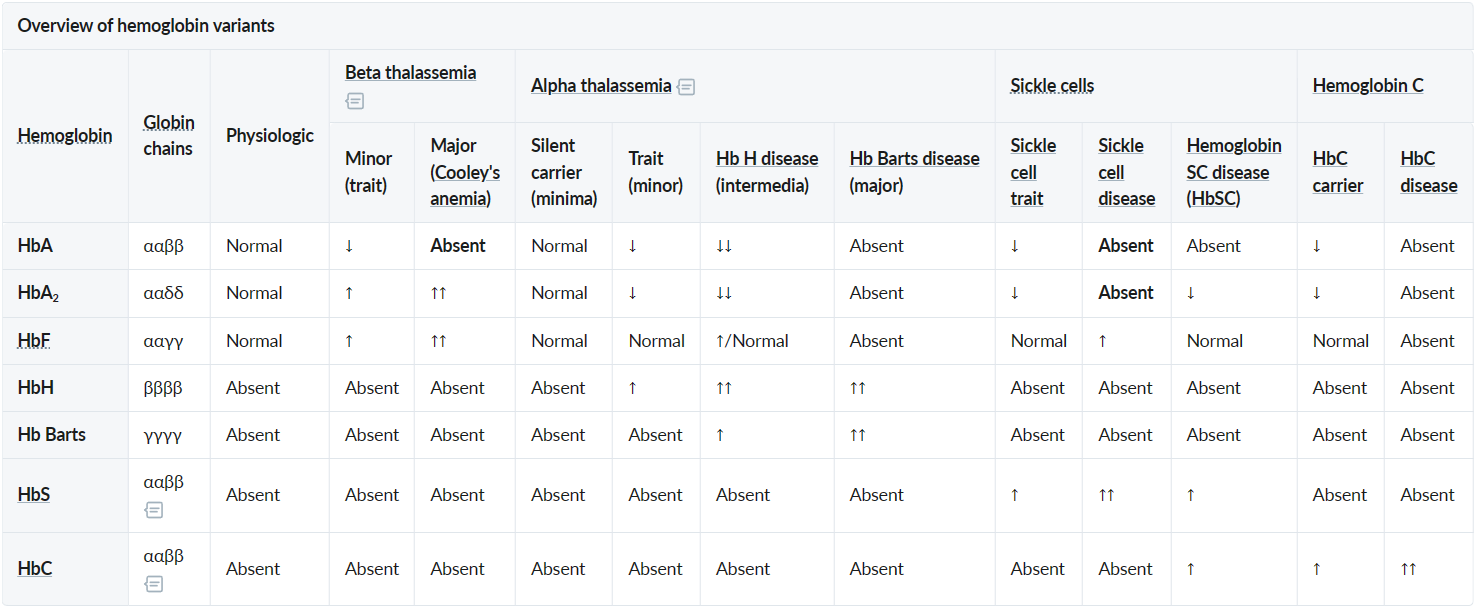
- Beta-thalassemia minor and major: faulty β-globin chain synthesis → ↓ β-chains→ ↑ γ-,δ-chains → ↑ HbF and ↑ HbA2.
- Alpha-thalassemia intermedia (HbH disease) and alpha-thalassemia major (Bart’s disease): faulty α-globin chain synthesis → ↓ α-chains → impaired pairing of α-chains with β-chains and γ-chains→ ↑ free β-, γ-chains → ↑ HbH, ↑ Hb Barts
- Sickle cell anemia: Point mutation in the β-globin gene (chromosome 11) → glutamic acid replaced with valine (single amino acid substitution) → 2 α-globin and 2 mutated β-globin subunits create pathological hemoglobin S (HbS).
- Increased hemolysis: One of the chains (either α or β) is reduced → compensatory overproduction of other chains → excess globin chains precipitate and form inclusions within RBCs → erythrocyte instability with hemolysis
- Anemia → ↑ erythropoietin → bone marrow hyperplasia and skeletal deformities
Tip
Hemoglobin A2 is elevated in beta-thalassemia to compensate for beta-globin–chain underproduction, but results in microcytic red cells that are prone to hemolysis. Increased erythrocyte turnover results in a misleadingly low hemoglobin A1c level. In such cases, measurement of glycated serum proteins (ie, fructosamine) is sometimes used to estimate glycemic control. See Hyperglycemia tests
Clinical features
- Alpha (α)-Thalassemia
- 1 gene deletion (Silent Carrier): Asymptomatic, no treatment needed.
- 2 gene deletions (α-Thalassemia Trait): Usually asymptomatic with mild microcytic anemia.
- 3 gene deletions (HbH Disease): Excess β-chains form HbH (β₄) tetramers. This leads to moderate-to-severe chronic hemolytic anemia and splenomegaly.
- 4 gene deletions (Hb Barts): Incompatible with life, resulting in hydrops fetalis. Excess γ-chains (from fetal Hb) form Hb Barts (γ₄).
- Mechanism: Hb Barts has an extremely high affinity for oxygen. It binds O2 in the placenta but fails to release it to fetal tissues.
- Result: This leads to profound tissue hypoxia, causing a cascade of:
- High-Output Heart Failure: The heart overworks to compensate for the lack of oxygen delivery.
- Massive Edema (Anasarca): The combination of heart failure and liver dysfunction (from extramedullary hematopoiesis) causes fluid to leak out of the vessels.
- Beta (β)-Thalassemia
- β-Thalassemia Minor (Trait): Heterozygous (one defective gene). Typically asymptomatic with mild microcytic anemia. A classic finding is a normal or increased RBC count despite low Hb and MCV.
- β-Thalassemia Major (Cooley Anemia): Homozygous (two defective genes). Severe anemia becomes symptomatic around 6 months of age as fetal hemoglobin (HbF) levels decline.
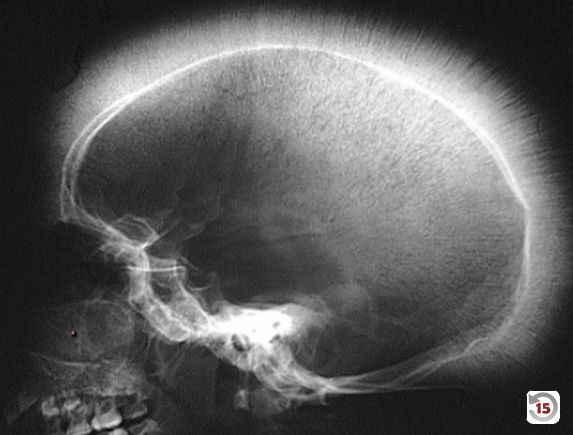
- Clinical Features: Massive hepatosplenomegaly from extramedullary hematopoiesis, “crewcut” appearance on skull x-ray, and “chipmunk” facies due to bone marrow expansion.
- Requires lifelong blood transfusions.
Diagnostics
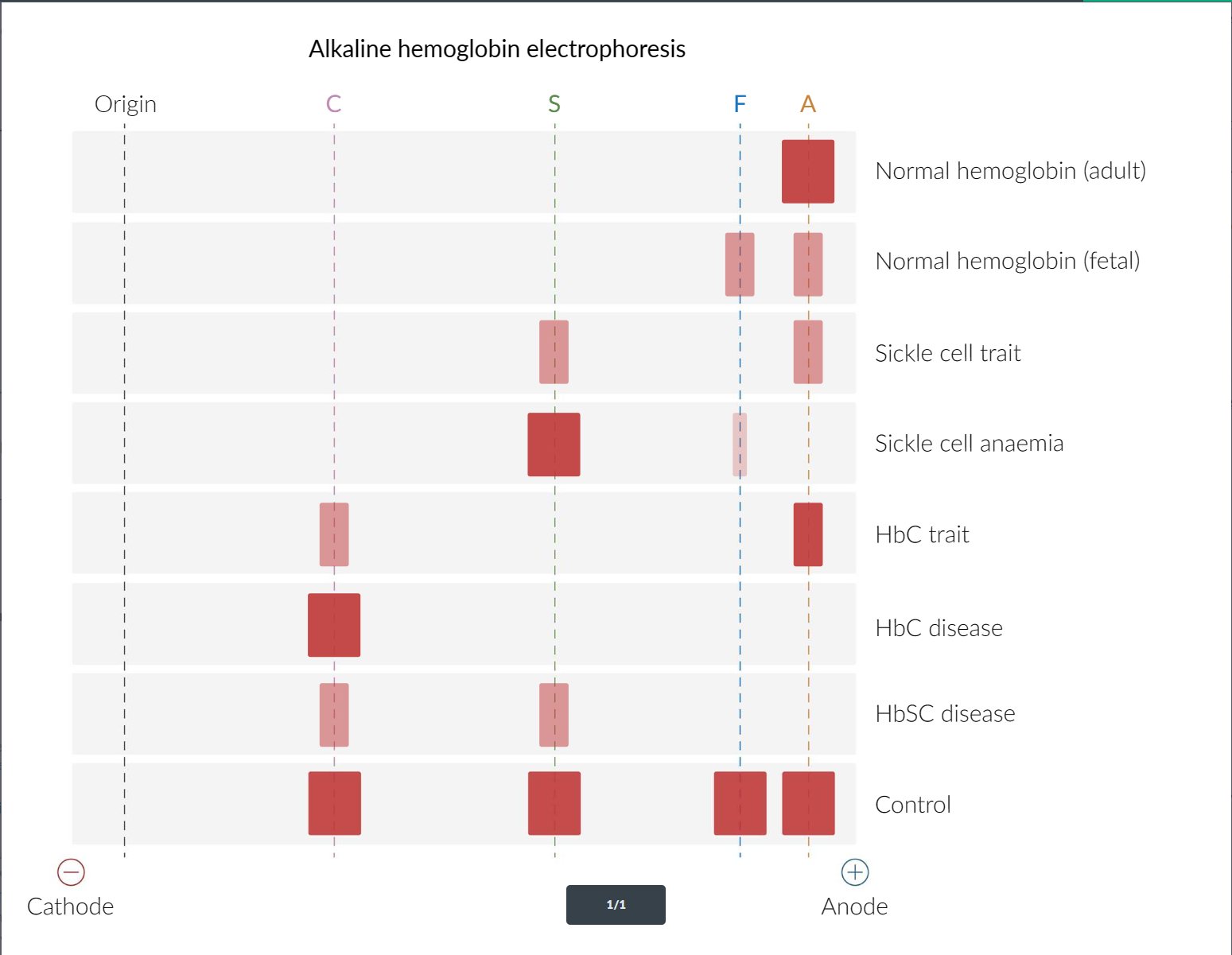
- Hb-electrophoresis
Tip
α-thalassemia minima vs β-thalassemia minor:
- Both asymptomatic
- α-thalassemia minima is normal on electrophoresis, while β-thalassemia shows narrowed HbA band and widened HbA2 band
- Peripheral blood smear findings include:
- Target cells
- ↑ surface area-to-volume ratio
- Teardrop cells
- Target cells
- Skull x-ray: Hair-on-end (also known as “crew cut”) sign, caused by excessive expansion of the bone marrow to compensate for the destruction of red blood cells