Definition: A group of disorders characterized by an impaired energy production that mainly affects organs with a high energy requirement (e.g., brain).
Epidemiology
Etiology
Caused by defects in mitochondrial DNA, which are maternally inherited
- Children of an affected mother will likewise be affected.
- Genetic expression is variable due to heteroplasmy.
Pathophysiology
- Impaired oxidative phosphorylation → decreased production of energy in mitochondria (lack of ATP) → up-regulation of glycolysis → overproduction of pyruvate → accumulation of lactate and alanine
- Organs with a high energy requirement (e.g., retina, brain, inner ear, skeletal, cardiac muscles) are particularly affected.
Clinical features
- Commonly external ophthalmoplegia, ptosis, and/or exertional muscle weakness.
Subtypes of mitochondrial myopathies
- MELAS: characterized by Mitochondrial Encephalomyopathy, Lactic Acidosis, recurring Stroke-like episodes
- Other findings include
- Muscle weakness
- Tonic-clonic seizures
- Other findings include
Diagnostics
- Genetic studies (including mitochondrial DNA)
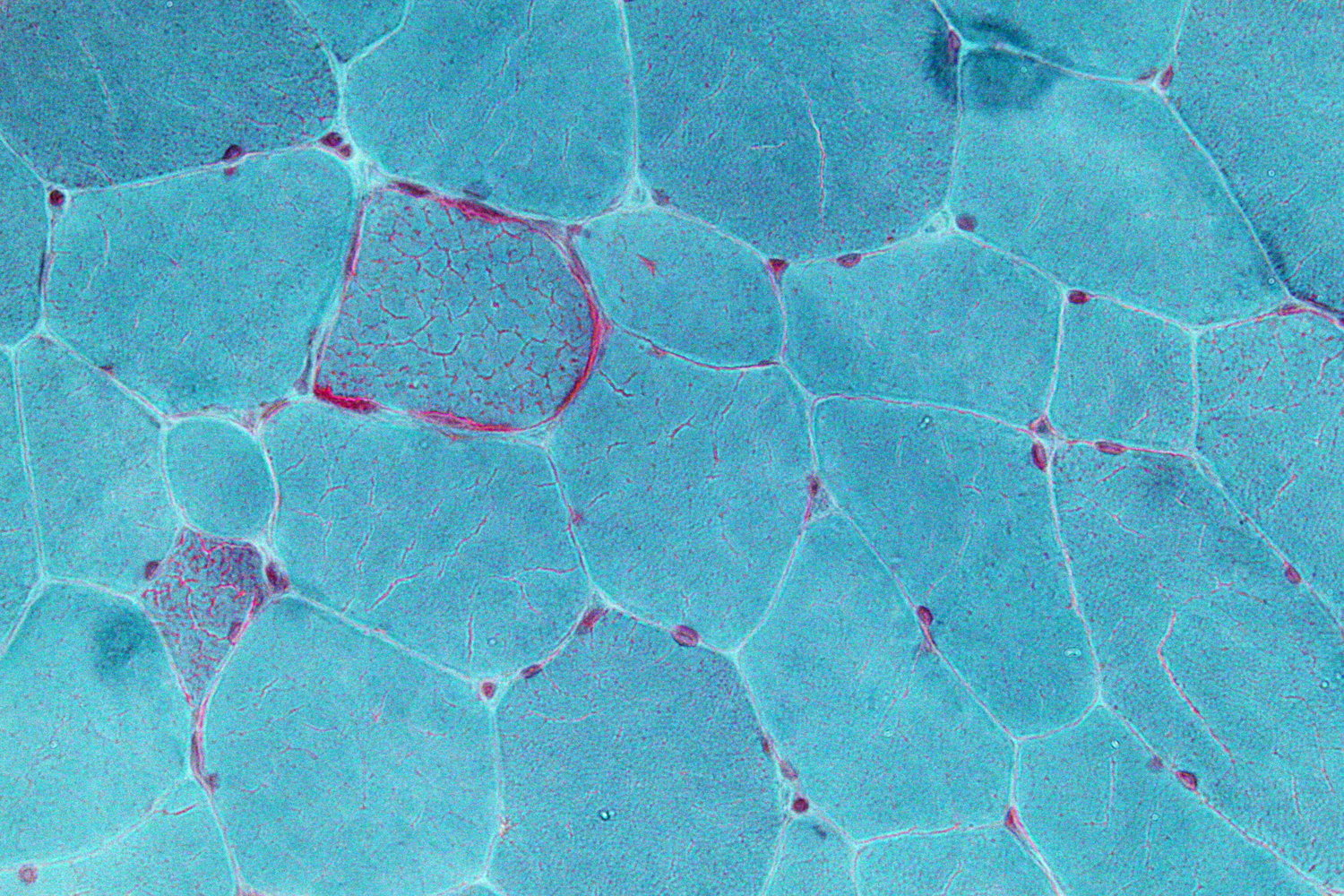
- Muscle biopsy: Immunohistochemistry typically shows ragged red fibers, which are caused by subsarcolemmal and intermyofibrillar accumulation of defective mitochondria in muscles (mitochondria stain red).
- Laboratory studies
- Normal CK
- Mitochondrial myopathies often cause mitochondrial dysfunction, which can lead to muscle weakness without significant muscle fiber death.
- Elevated lactate and alanine in serum, urine and/or CSF — lactic acidosis
- Normal CK