Epidemiology
Etiology
Pathophysiology
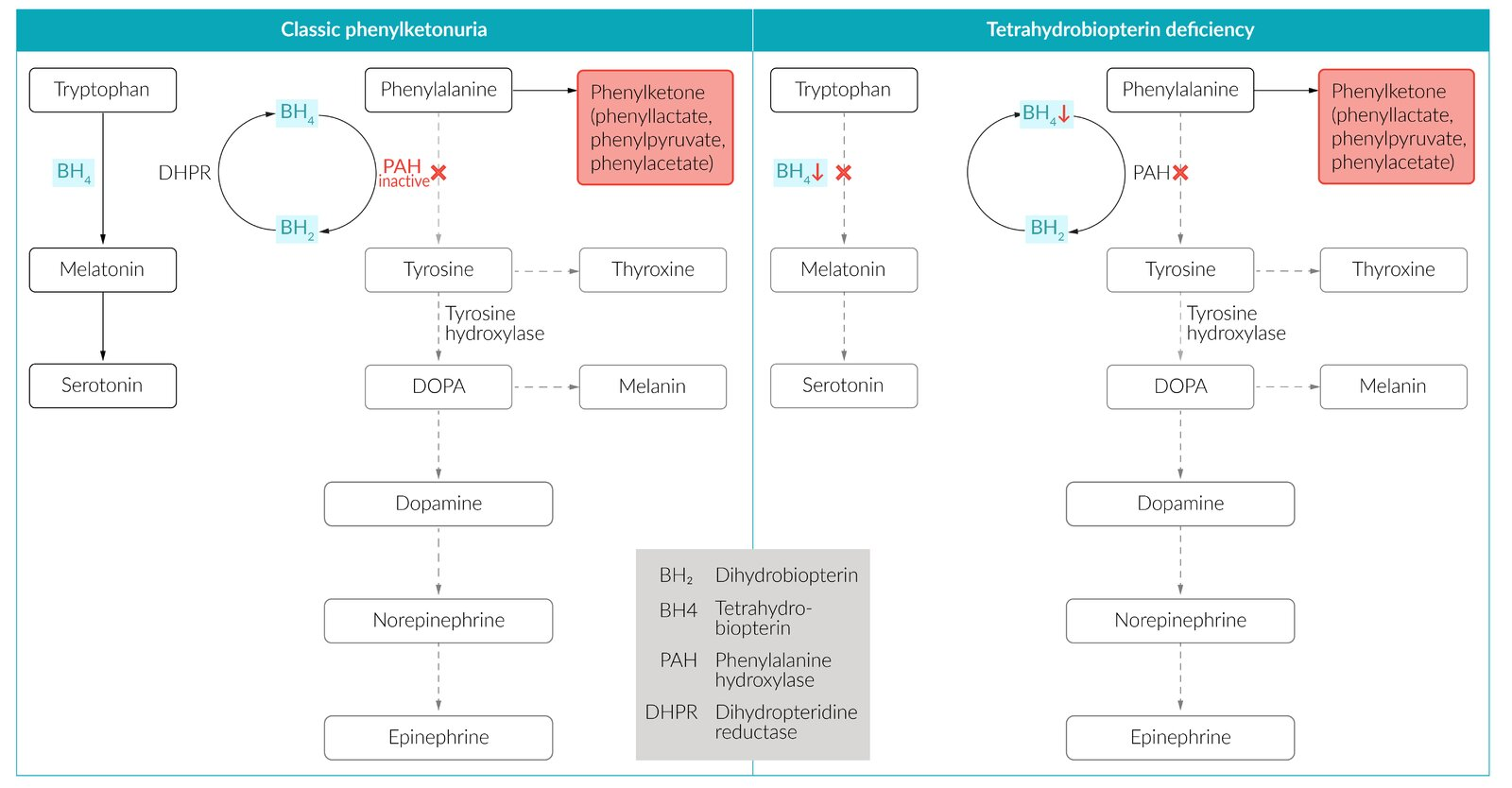
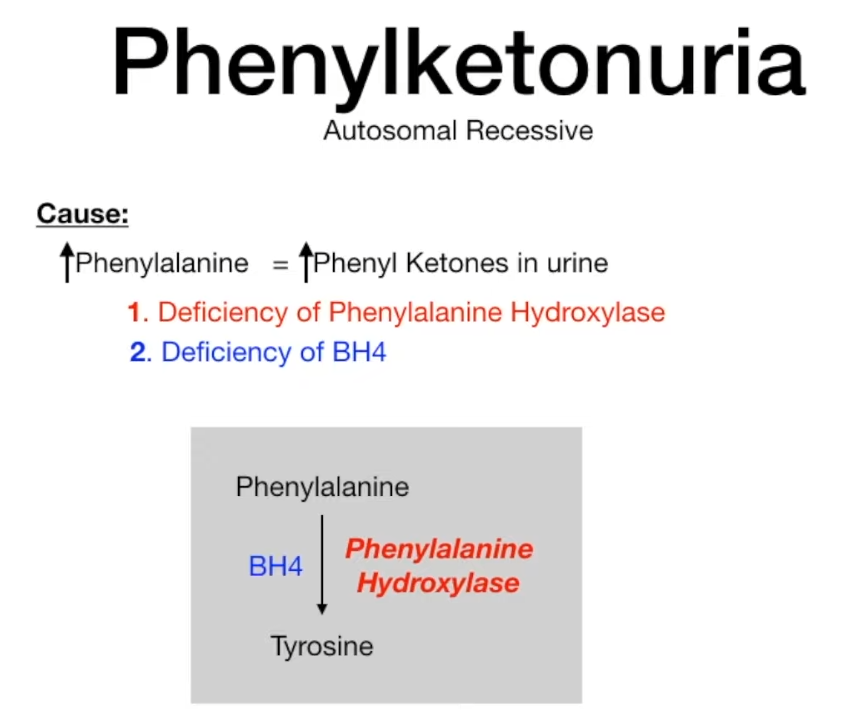
- Most commonly due to a defect of the liver enzyme phenylalanine hydroxylase (PAH) → impaired conversion of phenylalanine to tyrosine → tyrosine becomes nutritionally essential (classical PKU)
- Less commonly
- Tetrahydrobiopterin deficiency (malignant PKU): due to tetrahydrobiopterin deficiency (a cofactor of phenylalanine metabolism), caused by a deficiency in dihydropteridine reductase (normally reduces dihydrobiopterin to BH4), resulting in:
- Hyperphenylalaninemia due to ↓ conversion of phenylalanine to tyrosine → ↓ synthesis of catecholamines (BH4 is a cofactor for phenylalanine hydroxylase and tyrosine hydroxylase)
- ↓ Synthesis of serotonin (BH4 is a cofactor for tryptophan hydroxylase) → deficiencies of neurotransmitters
- Symptom severity varies between affected individuals.
- Tetrahydrobiopterin deficiency (malignant PKU): due to tetrahydrobiopterin deficiency (a cofactor of phenylalanine metabolism), caused by a deficiency in dihydropteridine reductase (normally reduces dihydrobiopterin to BH4), resulting in:
Clinical features
Phenylketonuria
 Link to original
Link to originalMnemonic
Musty → having a stale, moldy, or damp smell → MOLD-E

- Blue eyes, light skin, pale hair
- Due to a lack of melanin
Diagnostics
- Newborn Screening (Blood phenylalanine): Universal screening via heel prick 24-72 hours after birth. Measures Phe levels using tandem mass spectrometry. This is the primary screening test.
- Serum Amino Acid Analysis: The confirmatory test. Shows ↑ Phenylalanine and ↓ Tyrosine levels in the plasma, confirming the diagnosis and establishing the severity.
- Urinary Pteridine Analysis: Performed after a confirmed high Phe level to differentiate between PAH deficiency (classic PKU) and BH4 deficiency. Abnormal pterin profile indicates a defect in BH4 synthesis or regeneration.
- Urinary Ferric Chloride Test: A historical, now obsolete, test. Urine turns a blue-green color in the presence of phenylpyruvic acid. Not used for screening or diagnosis today.
Treatment
- Low phenylalanine and high tyrosine diet
- BH4 deficiency: supplementation of BH4 and possibly levodopa and 5-hydroxytryptophan